
Langerhans Cell Histiocytosis-Related PH Benefits from Advanced Therapies, According to Individual Case Study
Written by |
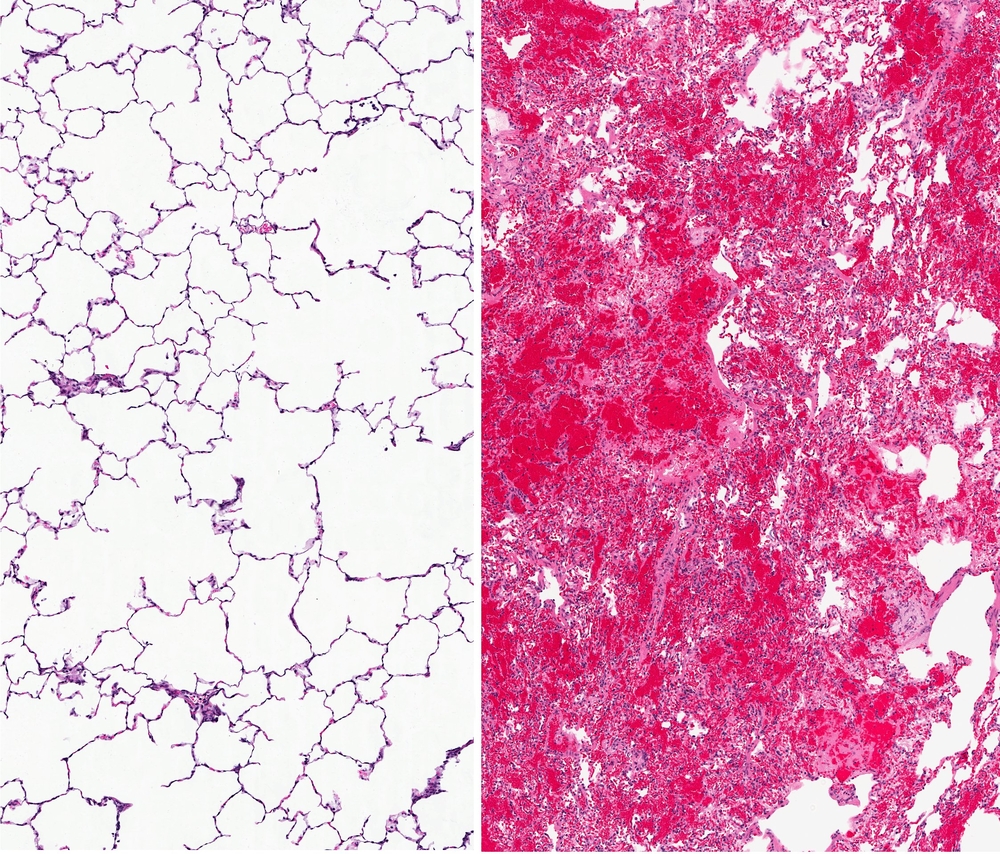
In a new case-report study entitled “Dramatic and sustained responsiveness of pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension to vasodilator therapy,” authors describe vasodilators’ positive effects in treating Pulmonary Langerhans cell histiocytosis patients with known pulmonary hypertension. The study was published in the journal Respiratory Medicine Case Reports.
Interstitial lung diseases are characterized by defects in the tissue surrounding the air sacs of the lungs that extends throughout both lungs, known as the interstitium.
[adrotate group=”4″]
Pulmonary Langerhans cell histiocytosis (PLCH) is a rare disease affecting mostly young smokers, where there is an accumulation of a specific type of antigen-presenting cells — the Langerhans’ cells — in the lungs’ small airways. A common complication of PLCH is pulmonary hypertension (PH), however, how advanced PH therapies impact PLCH-related PH is incompletely understood. In this study the authors describe the case of a PLCH patient with severe, disease-related PH that was submitted to advanced PH therapies.
The patient, a Caucasian woman, 46 years of age was diagnosed with Pulmonary Langerhans cell histiocytosis (PLCH) after a lung biopsy and a history of smoking (30 years) as well as mild, untreated obstructive sleep apnea (OSA). Additional tests revealed morphological alterations in the lung — the presence of bilateral cysts and scattered lung nodes (observed with high resolution computed tomography). Both Langerhans and other inflammatory cells composed the nodes and lung function tests showed mild impaired expiratory air-flow and gas-exchange capacity, while maintaining lung volumes. After an initial (and failed) treatment with prednisone and subsequent 2-Chlorodeox- yadenosine, the patient was then submitted to oral diltiazem for 6 months but with no significant improvements. An advanced therapy applied to pulmonary hypertension was introduced and the patient was treated with a combination of bosentan (endothelin receptor antagonist) followed by sildenafil (phosphodiesterase-5 inhibitor). The treatment was maintained for approximately seven years, at which point sildenafil was replaced by tadalafil.
[adrotate group=”3″]
The authors note that within ten years after initiation of advanced PH therapy, echocardiographic and functional tests improved significantly and were maintained, despite the patient’s continued smoking. The authors concluded that advanced PH therapies are an effective strategy for PLCH-related PH.


Leave a comment
Fill in the required fields to post. Your email address will not be published.