
Study Highlights Pulmonary Hypertension as a Result of Takayasu Arteritis
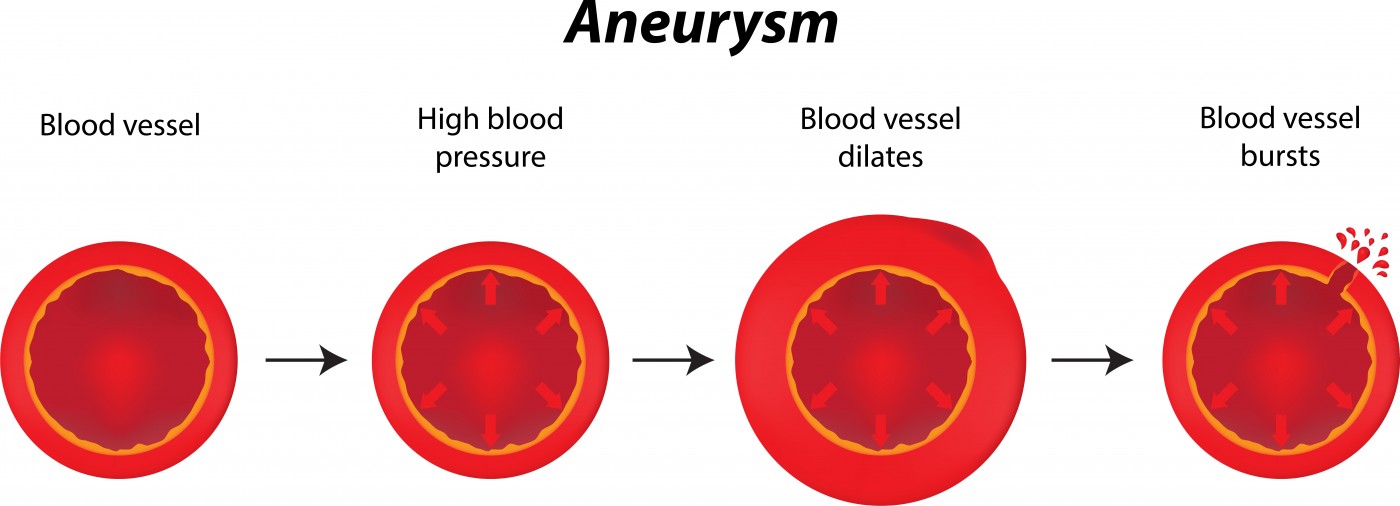



 Adding to the list of medical conditions that may result in pulmonary hypertension is the rare artery disease known as Takayasu arteritis. According to the Mayo Clinic, patients with Takayasu arteritis — which is grouped into a larger category of diseases that includes other types of vasculitis — have blood vessel inflammation, primarily in the aorta, that leads to stenosis (narrowed arteries) or aneurysms (abnormally dilated arteries). New research from Fuwai Hospital in China indicates that Takayasu arteritis can also lead to pulmonary hypertension.
Adding to the list of medical conditions that may result in pulmonary hypertension is the rare artery disease known as Takayasu arteritis. According to the Mayo Clinic, patients with Takayasu arteritis — which is grouped into a larger category of diseases that includes other types of vasculitis — have blood vessel inflammation, primarily in the aorta, that leads to stenosis (narrowed arteries) or aneurysms (abnormally dilated arteries). New research from Fuwai Hospital in China indicates that Takayasu arteritis can also lead to pulmonary hypertension.
Leading the effort were Drs. Xu Wang and Qing Liu, who published the results in The Journal of Rheumatology. During their study of “Takayasu Arteritis-associated Pulmonary Hypertension,” the objective was “to investigate the probable pathogenesis, clinical features, diagnosis, and therapy of patients with pulmonary hypertension in Takayasu arteritis.” To do so, the team found 98 patients with Takayasu arteritis with or without pulmonary hypertension and compared clinical features among three groups.
[adrotate group=”4″]
During the study, which was conducted between 2009 and 2013, the researchers examined Takayasu arteritis patients with pulmonary hypertension or idiopathic pulmonary arterial hypertension and Takayasu arteritis patients with pulmonary arterial involvement but no pulmonary hypertension. Some patients also presented with left heart disease. Patient symptoms, including those due to unexplained causes, were treated appropriately in the clinic, and clinicians conducted follow-ups to determine correlations between improvements and patient characteristics.
The researchers identified that Takayasu arteritis patients are likely to develop pulmonary hypertension. Pulmonary arterial systolic pressure was positively correlated with endothelin 1, a serum biomarker of pulmonary hypertension.
Patients with pulmonary hypertension due to pulmonary arterial involvement, rather than idiopathic pulmonary arterial hypertension, exhibited lower average cardiac indexes during right heart catheterization. All of the pulmonary arterial involvement patients responded favorably to acute vasodilator testing, compared to only 33.3% of the idiopathic pulmonary hypertension patients. However, three pulmonary arterial involvement patients died of heart failure during the follow-up period.
[adrotate group=”3″]
For six patients who underwent pulmonary artery revascularization as a means to treat symptoms, prognoses were good during the follow-up period, which was an average of 6.2 months following treatment. The twelve patients who had left heart disease also showed a favorable prognosis during the follow-up period, with one death due to heart failure.
Overall, the researchers believe early screening for pulmonary hypertension is important for patients with Takayasu arteritis. Even unexplained symptoms are important to identify and treat in order to increase the chances of patient survival. It seems as though revascularization and other pulmonary hypertension-specific therapies are effective treatments for those patients with pulmonary arterial involvement, pulmonary hypertension, and severe pulmonary artery stenosis.



